
En un escenario de pandemia vírica como en la que estamos inmersos, tener una idea clara de las cepas del virus que están circulando es fundamental. Para ello, tenemos que secuenciar el mayor número de cepas para que tener una imagen lo más clara posible de cuál es la situación. Eso significa dinero y un reto tecnológico.

Cuando secuencié mi primer gen, que publicamos en 1996, la tecnología era bastante primitiva. Hoy en día, aquello con lo que peleábamos en los laboratorios de investigación está pulido y afinado por algunas empresas que han conseguido estandarizar, optimizar y abaratar el proceso.
El primer paso que tenemos que hacer para secuenciar un virus como Sars-CoV-2 es retrotranscribir su ARN en ADN. El ADN es una molécula más estable y más fácil de operar.

En el diagrama abajo se puede apreciar todos los pasos que tenemos que seguir para llegar a tener el genoma de las cepas de Sars-CoV-2 que vamos a secuenciar.
¿Cómo conseguimos que la muestra de un pocillo de una placa de 96 esté correctamente identificada?


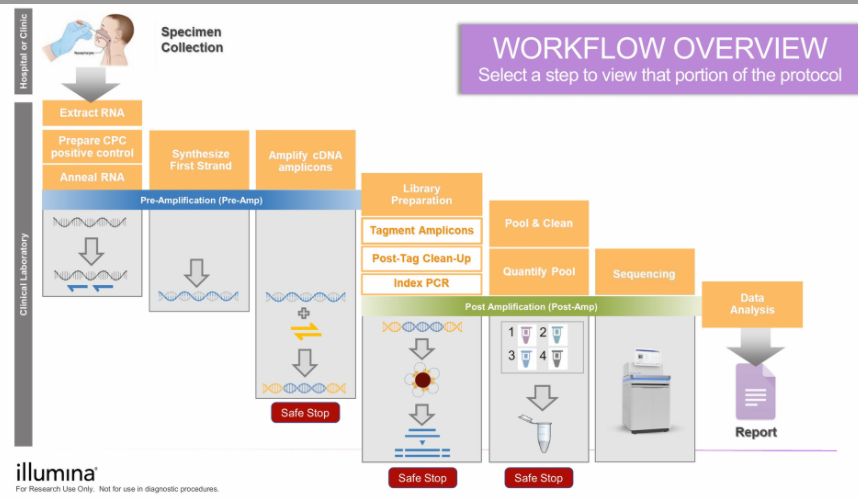
Hay mucha bioquímica y biología molecular en todo el proceso. Intentaré resumirlo. Los trucos que utilizan, en este caso los científicos e ingenieros, se basan en tener millones de copias de cada uno de los fragmentos que vamos a secuenciar, tener los fragmentos identificados con secuencias características de ADN característicos, la capacidad de unir el ADN a perlas magnéticas que nos permiten limpiar el ADN y seleccionar fragmentos de una longitud determinada. Al tener muchísimas copias de cada fragmento, cuando se secuencien, usando la técnica de nucleóticos que al unirse al fragmento que se están copiando y emiten luz, podemos descartar aquellas emisiones de luz que son poco claras ¿Por qué? por que tenemos millones de lecturas y podemos darnos el lujo de eliminar aquellas que no tengan un nivel de calidad aceptable.
¿Cómo conseguimos que la muestra de un pocillo de una placa de 96 esté correctamente identificada?
Gracias a los UDI (Unique dual indexes) podemos tener secuencias de índice distintas y no relacionadas para cada una de las lecturas de índice i5 e i7 en cada uno de los pocillos de una placa de 96 pocillos.

Se trata de una placa de 96 pocillos en los que los UDIs ya están precargados, así que cuando pones la muestra y esos fragmentos se incorporan a las secuencias que quieres secuenciar, los UDIs se incorporan a los fragmentos identificándolos a cada uno de los pocillos. De esa manera podemos tener las 96 muestras (incluidos los controles positivos y negativos) identificadas correctamente ¿Cuál es la ventaja? que podemos ahora meter las 96 muestras en un solo tuvo ¿Qué significa eso? que no tenemos que pipetear en los 96 pocillos, ahora trabajamos ¡Con un solo tubo! quien sabe lo que eso significa le hace la ola a los que diseñaron este método.
Perlas magnéticas limpian y estandarizan la longitud de nuestros fragmentos


Secuenciación por Síntesis (Illumina): Conceptos Básicos. Fuente





















